Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 2008, 9(559):1-13.
#WGCNA 包通过 bioconductor 安装
#install.packages('BiocManager')
#BiocManager::install('WGCNA')
library(WGCNA)
#示例数据,基因表达值矩阵
gene <- read.delim('gene_express.txt', row.names = 1, check.names = FALSE)
#例如过滤低表达值的基因,只保留平均表达值在 1 以上的
gene <- subset(gene, rowSums(gene)/ncol(gene) >= 1)
#转置矩阵,行是基因,列是样本
gene <- t(gene)

##power 值选择
powers <- 1:20
sft <- pickSoftThreshold(gene, powerVector = powers, verbose = 5)
#拟合指数与 power 值散点图
par(mfrow = c(1, 2))
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2], type = 'n',
xlab = 'Soft Threshold (power)', ylab = 'Scale Free Topology Model Fit,signed R^2',
main = paste('Scale independence'))
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels = powers, col = 'red');
abline(h = 0.90, col = 'red')
#平均连通性与 power 值散点图
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab = 'Soft Threshold (power)', ylab = 'Mean Connectivity', type = 'n',
main = paste('Mean connectivity'))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels = powers, col = 'red')


#上一步估计的最佳 power 值
powers <- sft$powerEstimate
#获得 TOM 矩阵
adjacency <- adjacency(gene, power = powers)
tom_sim <- TOMsimilarity(adjacency)
rownames(tom_sim) <- rownames(adjacency)
colnames(tom_sim) <- colnames(adjacency)
tom_sim[1:6,1:6]
#输出 TOM 矩阵
#write.table(tom_sim, 'TOMsimilarity.txt', sep = '\t', col.names = NA, quote = FALSE)

#TOM 相异度 = 1 – TOM 相似度
tom_dis <- 1 - tom_sim
#层次聚类树,使用中值的非权重成对组法的平均聚合聚类
geneTree <- hclust(as.dist(tom_dis), method = 'average')
plot(geneTree, xlab = '', sub = '', main = 'Gene clustering on TOM-based dissimilarity',
labels = FALSE, hang = 0.04)

#使用动态剪切树挖掘模块
minModuleSize <- 30 #模块基因数目
dynamicMods <- cutreeDynamic(dendro = geneTree, distM = tom_dis,
deepSplit = 2, pamRespectsDendro = FALSE, minClusterSize = minModuleSize)
table(dynamicMods)

#模块颜色指代
dynamicColors <- labels2colors(dynamicMods)
table(dynamicColors)
plotDendroAndColors(geneTree, dynamicColors, 'Dynamic Tree Cut',
dendroLabels = FALSE, addGuide = TRUE, hang = 0.03, guideHang = 0.05,
main = 'Gene dendrogram and module colors')

#基因表达聚类树和共表达拓扑热图
plot_sim <- -(1-tom_sim)
#plot_sim <- log(tom_sim)
diag(plot_sim) <- NA
TOMplot(plot_sim, geneTree, dynamicColors,
main = 'Network heatmap plot, selected genes')

#计算基因表达矩阵中模块的特征基因(第一主成分)
MEList <- moduleEigengenes(gene, colors = dynamicColors)
MEs <- MEList$eigengenes
head(MEs)[1:6]
#输出模块特征基因矩阵
#write.table(MEs, 'moduleEigengenes.txt', sep = '\t', col.names = NA, quote = FALSE)

#通过模块特征基因计算模块间相关性,表征模块间相似度
ME_cor <- cor(MEs)
ME_cor[1:6,1:6]
#绘制聚类树观察
METree <- hclust(as.dist(1-ME_cor), method = 'average')
plot(METree, main = 'Clustering of module eigengenes', xlab = '', sub = '')
#探索性分析,观察模块间的相似性
#height 值可代表模块间的相异度,并确定一个合适的阈值作为剪切高度
#以便为低相异度(高相似度)的模块合并提供依据
abline(h = 0.2, col = 'blue')
abline(h = 0.25, col = 'red')
#模块特征基因聚类树热图
plotEigengeneNetworks(MEs, '', cex.lab = 0.8, xLabelsAngle= 90,
marDendro = c(0, 4, 1, 2), marHeatmap = c(3, 4, 1, 2))

#相似模块合并,以 0.25 作为合并阈值(剪切高度),在此高度下的模块将合并
#近似理解为相关程度高于 0.75 的模块将合并到一起
merge_module <- mergeCloseModules(gene, dynamicColors, cutHeight = 0.25, verbose = 3)
mergedColors <- merge_module$colors
table(mergedColors)
#基因表达和模块聚类树
plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors), c('Dynamic Tree Cut', 'Merged dynamic'),
dendroLabels = FALSE, addGuide = TRUE, hang = 0.03, guideHang = 0.05)

#患者的临床表型数据
trait <- read.delim('trait.txt', row.names = 1, check.names = FALSE)
#使用上一步新组合的共表达模块的结果
module <- merge_module$newMEs
#患者基因共表达模块和临床表型的相关性分析
moduleTraitCor <- cor(module, trait, use = 'p')
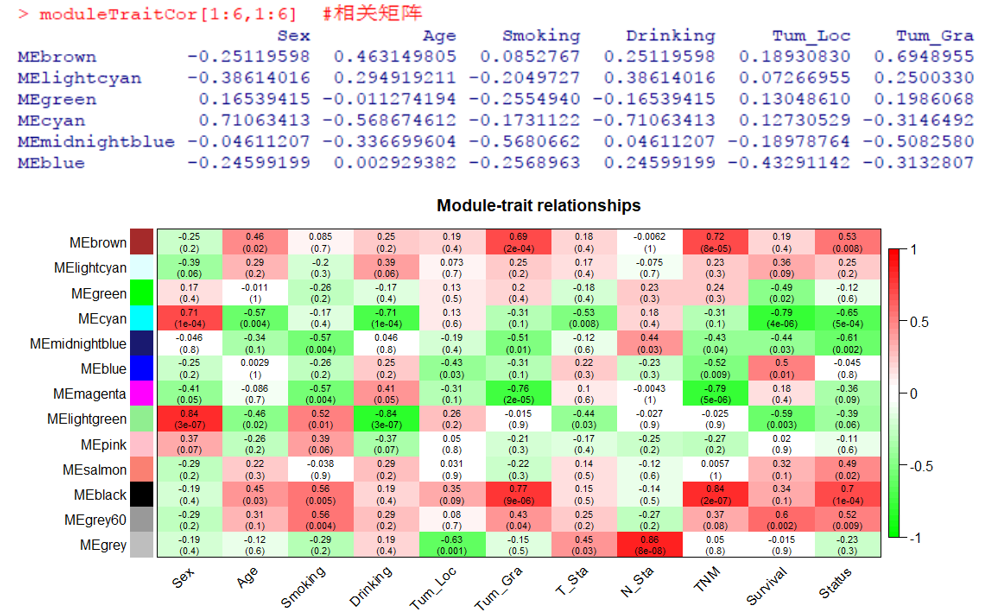
moduleTraitCor[1:6,1:6] #相关矩阵
#相关系数的 p 值矩阵
moduleTraitPvalue <- corPvalueStudent(moduleTraitCor, nrow(module))
#输出相关系数矩阵或 p 值矩阵
#write.table(moduleTraitCor, 'moduleTraitCor.txt', sep = '\t', col.names = NA, quote = FALSE)
#write.table(moduleTraitPvalue, 'moduleTraitPvalue.txt', sep = '\t', col.names = NA, quote = FALSE)
#相关图绘制
textMatrix <- paste(signif(moduleTraitCor, 2), '\n(', signif(moduleTraitPvalue, 1), ')', sep = '')
dim(textMatrix) <- dim(moduleTraitCor)
par(mar = c(5, 10, 3, 3))
labeledHeatmap(Matrix = moduleTraitCor, main = paste('Module-trait relationships'),
xLabels = names(trait), yLabels = names(module), ySymbols = names(module),
colorLabels = FALSE, colors = greenWhiteRed(50), cex.text = 0.7, zlim = c(-1,1),
textMatrix = textMatrix, setStdMargins = FALSE)
#基因与模块的对应关系列表
gene_module <- data.frame(gene_name = colnames(gene), module = mergedColors, stringsAsFactors = FALSE)
head(gene_module)
#“black”模块内的基因名称
gene_module_select <- subset(gene_module, module == 'black')$gene_name
#“black”模块内基因在各样本中的表达值矩阵(基因表达值矩阵的一个子集)
gene_select <- t(gene[,gene_module_select])
#“black”模块内基因的共表达相似度(在 TOM 矩阵中提取子集)
tom_select <- tom_sim[gene_module_select,gene_module_select]
#输出
#write.table(gene_select, 'gene_select.txt', sep = '\t', col.names = NA, quote = FALSE)
#write.table(tom_select, 'tom_select.txt', sep = '\t', col.names = NA, quote = FALSE)

#选择 black 模块内的基因
gene_black <- gene[ ,gene_module_select]
#基因的模块成员度(module membership)计算
#即各基因表达值与相应模块特征基因的相关性,其衡量了基因在全局网络中的位置
geneModuleMembership <- signedKME(gene_black, module['MEblack'], outputColumnName = 'MM')
MMPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nrow(module)))
#各基因表达值与临床表型的相关性分析
geneTraitSignificance <- as.data.frame(cor(gene_black, trait['TNM'], use = 'p'))
GSPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nrow(trait)))
#选择显著(p<0.01)、高 black 模块成员度(MM>=0.8),与 TNM 表型高度相关(r>=0.8)的基因
geneModuleMembership[geneModuleMembership<0.8 | MMPvalue>0.01] <- 0
geneTraitSignificance[geneTraitSignificance<0.8 | MMPvalue>0.01] <- 0
select <- cbind(geneModuleMembership, geneTraitSignificance)
select <- subset(select, geneModuleMembership>=0.8 & geneTraitSignificance>=0.8)
head(select)
#候选基因与 TNM 分期的相关性散点图
dir.create('black_TNM_cor', recursive = TRUE)
for (i in rownames(select)) {
png(paste('black_TNM_cor/', i, '-TNM.png', sep = ''),
width = 4, height = 4, res = 400, units = 'in', type = 'cairo')
plot(trait[ ,'TNM'], gene[ ,i], xlab = 'TNM', ylab = i,
main = paste('MM_black = ', select[i,'MMblack'], '\ncor_TNM = ', select[i,'TNM']), cex = 0.8, pch = 20)
fit <- lm(gene[ ,i]~trait[ ,'TNM'])
lines(trait[ ,'TNM'], fitted(fit))
dev.off()
}
#候选基因间的 TOM 矩阵
plotNetworkHeatmap(gene,
plotGenes = rownames(select),
networkType = 'unsigned',
useTOM = TRUE,
power = powers,
main = 'unsigned correlations')

#输出各模块子网络的边列表和节点列表,后续可导入 cytoscape 绘制网络图
#其中,边的权重为 TOM 矩阵中的值,记录了基因间共表达相似性
dir.create('cytoscape', recursive = TRUE)
for (mod in 1:nrow(table(mergedColors))) {
modules <- names(table(mergedColors))[mod]
probes <- colnames(gene)
inModule <- (mergedColors == modules)
modProbes <- probes[inModule]
modGenes <- modProbes
modtom_sim <- tom_sim[inModule, inModule]
dimnames(modtom_sim) <- list(modProbes, modProbes)
outEdge <- paste('cytoscape/', modules, '.edge_list.txt',sep = '')
outNode <- paste('cytoscape/', modules, '.node_list.txt', sep = '')
exportNetworkToCytoscape(modtom_sim,
edgeFile = outEdge,
nodeFile = outNode,
weighted = TRUE,
threshold = 0.3, #该参数可控制输出的边数量,过滤低权重的边
nodeNames = modProbes,
altNodeNames = modGenes,
nodeAttr = mergedColors[inModule])
}
